Fabrication décentralisée de médicaments en thérapies avancées

Clara Caja. Technicienne des affaires réglementaires chez Konexio Biotech
Par définition, les médicaments de thérapie innovante (ATMP) sont divisés en quatre grandes catégories, comme indiqué dans le règlement (CE) n° 1394/2007 et le décret législatif royal 1/2015 du 24 juillet : médicaments de thérapie génique, médicaments de thérapie cellulaire, produits d’ingénierie tissulaire et médicaments de thérapie innovante combinés à un ou plusieurs produits sanitaires.
Les médicaments de thérapie innovante sont régis par le règlement (CE) n° 1394/2007. Comme tous les médicaments, il est essentiel qu’ils soient fabriqués conformément aux bonnes pratiques de fabrication (BPF). Le volume 4 d’EudraLex (normes régissant les médicaments dans l’Union européenne) contient les lignes directrices pour l’interprétation des principes et des directives de bonnes pratiques de fabrication des médicaments à usage humain et vétérinaire.
Le volume 4 d’EudraLex est divisé en quatre parties : Partie I Exigences de base pour les médicaments, Partie II Exigences de base pour les substances actives utilisées comme matières premières, Partie III Documents relatifs aux BPF et Partie IV Exigences des BPF pour les médicaments de thérapie innovante. Par conséquent, les médicaments de thérapie innovante seront fabriqués conformément aux dispositions de la partie IV des NCF.
Il existe actuellement deux voies générales pour la livraison à grande échelle d’ATMP aux patients : la fabrication centralisée et la fabrication décentralisée (Figure 1).
La fabrication centralisée est considérée comme la fabrication traditionnelle de ces médicaments. Elle consiste en une installation unique qui assure la production et la fourniture des ATMP dans une vaste région géographique. Pour le traitement personnalisé, cela peut se produire dans une région discrète ou nécessiter le transport des cellules du patient sur de longues distances.
La fabrication décentralisée est apparue récemment et consiste à produire des ATMP dans des centres régionaux (« hubs ») proches des centres de traitement et à livrer les produits dans leur environnement immédiat.
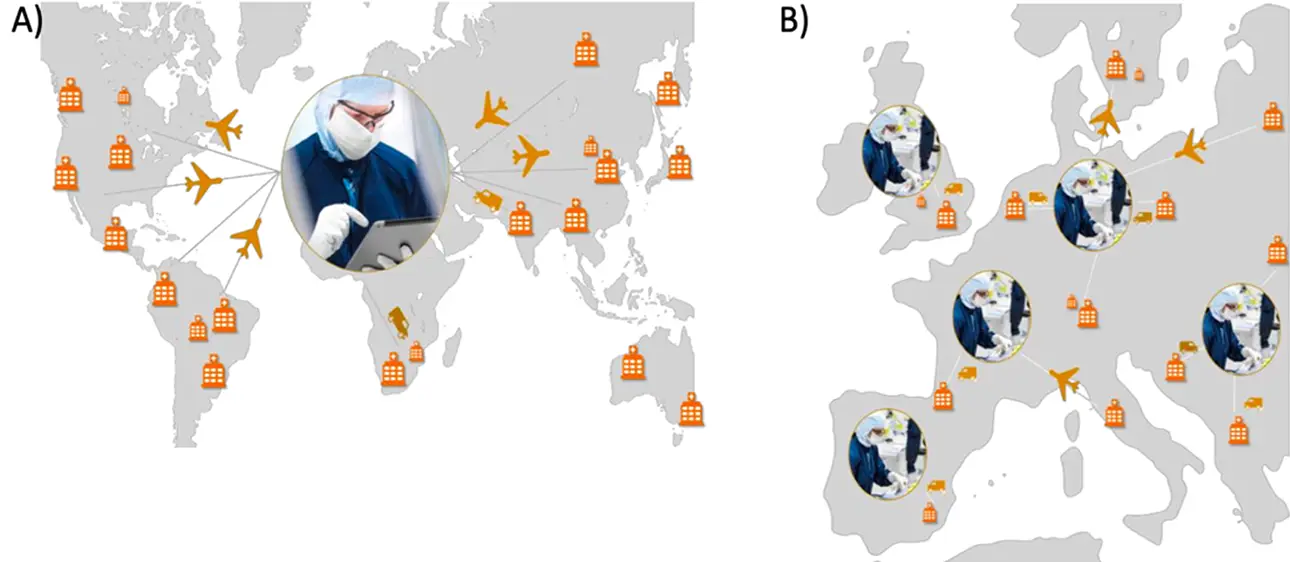
Figure 1. A) Fabrication centralisée d’ATMP. B) Fabrication décentralisée d’ATMP. Source : Feuille de route des besoins et de la stratégie de mise en œuvre des nouvelles thérapies innovantes. Horizon de restauration
La fabrication centralisée peut présenter certains inconvénients et défis par rapport à la fabrication décentralisée d’ATMP. La fabrication de médicaments conventionnels est un processus hautement centralisé et implique une logistique complexe de la chaîne d’approvisionnement et de la cryoconservation. Bien qu’il s’agisse d’un processus éprouvé dans le temps et économiquement viable pour les médicaments traditionnels, il peut être difficile à adapter aux médicaments de thérapie avancée, en particulier aux thérapies cellulaires autologues.
La fabrication centralisée de thérapies cellulaires peut prendre beaucoup de temps en raison de plusieurs facteurs. Tout d’abord, le processus d’isolement et d’expansion des cellules peut prendre beaucoup de temps. En outre, garantir l’accessibilité de la thérapie aux patients peut être une tâche longue et difficile, en particulier pour ceux qui sont éloignés des installations de fabrication centralisées. La logistique impliquée dans le transport de la thérapie vers ces patients peut poser des obstacles et entraîner des retards importants.
La décentralisation transforme la fabrication de la thérapie cellulaire en un processus plus distribué en la rapprochant des patients, car elle améliore l’accès et le confort du patient. Les médecins ayant accès à un laboratoire local de thérapie cellulaire et génique peuvent travailler avec le personnel local pour créer une approbation préalable, programmer le prélèvement, créer le traitement, libérer le traitement et administrer le traitement le plus rapidement possible. Les facteurs limitant le taux sont réduits car le matériel du patient est disponible localement pour le centre de traitement décentralisé, ce qui réduit les défis logistiques et de transport.
La fabrication décentralisée est le modèle préféré pour les produits cellulaires autologues. Il s’applique aussi bien aux approches pharmaceutiques qu’aux petites entreprises de biotechnologie et aux hôpitaux spécialisés qui produisent des ATMP. Chaque centre doit être en mesure de fournir des ATMP équivalents, quels que soient le lieu ou les opérateurs. C’est pourquoi des systèmes de gestion intégrés et automatisés ont été mis en place pour permettre au produit d’aphérèse de passer par les multiples étapes de fabrication dans un kit jetable à usage unique qui ne s’ouvre pas directement sur l’environnement. L’extraction d’échantillons et l’ajout de réactifs ou de milieux sont effectués à l’aide de soudeuses de tubes stériles ou d’orifices d’accès aseptiques, ce qui permet de maintenir ce que l’on appelle un système fonctionnellement fermé et de réduire ainsi les variations causées par les opérateurs. Les dispositifs automatisés sont devenus la méthode préférée pour la fabrication décentralisée de CAR-T. Certains de ces dispositifs sont CliniMACS Prodigy© ou Lonza Cocoon©.
Comme indiqué dans la partie IV des NCF, la certification et la libération des lots d’ATMP fabriqués dans un système décentralisé sont particulièrement importantes car la fabrication sur plusieurs sites augmente le risque de variabilité du produit.
En particulier, le processus de certification et de libération des lots doit garantir que chaque lot libéré sur l’un des sites a été fabriqué et vérifié conformément aux exigences d’autorisation de mise sur le marché/d’essai clinique et aux autres exigences réglementaires pertinentes, y compris le respect des NCF. Compte tenu de ce qui précède, les aspects suivants doivent être pris en compte :
Une installation centrale doit être identifiée, qui doit être établie dans l’UE. L’installation centrale est responsable de la supervision des sites décentralisés. L’installation centrale doit assumer au minimum les tâches suivantes : s’assurer que les personnes participant au processus de certification et de libération des lots sont dûment qualifiées et formées pour leurs tâches, et réaliser des audits pour confirmer que le processus de certification et de libération des lots est conforme à ce qui est décrit dans la procédure normalisée de travail (PNT).
Un contrat ou un accord technique doit être établi entre l’installation centrale et les sites décentralisés. Cet accord doit définir les responsabilités de chaque partie, y compris la responsabilité de la personne qualifiée (QP).
La personne qualifiée (QP) a la responsabilité ultime de certifier qu’un lot d’ATMP est conforme aux exigences de qualité et peut être libéré. La QP de l’installation centrale peut se fier aux données des sites décentralisés à condition qu’elles aient été fournies par du personnel qualifié et formé.
Pour toutes ces raisons, la fabrication décentralisée de médicaments de thérapie innovante, en particulier de thérapies cellulaires, représente un pas vers un système de soins de santé plus inclusif, où les traitements innovants ne sont pas un privilège, mais un droit accessible à tous.
Bibliographie
- Règlement (CE) n° 1394/2007 du Parlement européen et du Conseil du 13 novembre 2007 concernant les médicaments de thérapie innovante et modifiant la directive 2001/83/CE ainsi que le règlement (CE) n° 726/2004.
- Partie IV Directives sur les normes de bonne fabrication spécifiques aux médicaments de thérapie innovante.
- D 4.1 Roadmap of WP4. Roadmap of the needs and strategy of the implementation of new Advanced Therapies de Restore Horizon
- Shah, M., Krull, A., Odonnell, L., de Lima, M. J., & Bezerra, E. (2023). Promises and challenges of a decentralized CAR T-cell manufacturing model. Frontiers in transplantation, 2, 1238535.
- Decentralized manufacturing of cell and gene therapies: Overcoming challenges and identifying opportunities Harrison, Richard P. et al. Cytotherapy, Volume 19, Issue 10, 1140 – 1151
- Beyond Urban Centers: Expanding CAR-T Therapy with Decentralized Manufacturing. Boston Labs – Inspired Clinical Logistics.
Lundi - Vendredi: 9:00 - 19:00 h.
Newsletter
© 2024. Tous droits réservés.
Un autre site Web créé par Palabra de Ciervo